
G.Patton
Expert
- Joined
- Jul 5, 2021
- Messages
- 2,991
- Solutions
- 3
- Reaction score
- 3,378
- Points
- 113
- Deals
- 1
Wprowadzenie
Kwas lizergowy, podstawowy fragment pochodzący z alkaloidów sporyszu, został zsyntetyzowany w czternastu sekwencjach rozpoczynających się od 3-beta-karboksyetyloindolu. Materiał wyjściowy został przekształcony w półprodukt 1-benzoilo-5-keto-1,2,2a-3,4,5-heksahydrobenz-[cd]-indol (3), zawierający trzy z czterech pierścieni obecnych w kwasie lizergowym. Ten keton z kolei został przekształcony w związek tetracykliczny, 9-keto-7-metylo-4,5,5a,6,6a,7,8,9-oktahydroindolo-[4.3-fe]-chinolinę (8 ), a następnie w kwas liz ergowy (14). Synteza ta nie jest prosta i wymaga dużego doświadczenia laboratoryjnego i wiedzy chemicznej. Co więcej, istnieje kilka manipulacji z niebezpiecznymi substancjami, które muszą być przeprowadzane z zachowaniem ścisłych środków bezpieczeństwa.
Temperatura wrzenia: 536,2±50,0 °C przy 760 mm Hg;
Temperatura topnienia: 240 °C;
Masa cząsteczkowa: 268,31 g/mol;
Gęstość: 1,4 ± 0,1 g/mL;
Numer CAS: 82-58-6.
Sprzęt i szkło:
- Stalowy reaktor uwodorniający 2-3 L;
- Autoklaw stalowy 500 ml;
- Waga laboratoryjna (odpowiednia 0,01 - 500 g);
- Kolby okrągłodenne 100, 200, 500 ml, 5 i 10 l;
- Sprężarka i źródło wodoru (H2);
- Kolba Buchnera i lejek (duży) 5 L [filtr Schotta może być używany do małych ilości];
- Maszyna Rotovap (duża);
- Źródło próżni;
- Lejki rozdzielające 500 ml i 2 l;
- Balon z azotem ~50-70 l (1 bar);
- Zatyczki do kolb;
- Łaźnia z soloną wodą lodową;
- 5 L x2, 2 L x2; 1 L x2; 500 mL x2; 100 mL x3; 50 mL x2 Zlewki;
- Szklana strzykawka lub pipeta Pasteura;
- Mieszadło magnetyczne lub mieszadło górne;
- Zestaw do destylacji próżniowej;
- Chłodnica zwrotna;
- Statyw retortowy i zacisk do mocowania aparatury;
- Termometr laboratoryjny (od -20 °C do 200 °C) z adapterem do kolby;
- Papier wskaźnikowy pH;
- Szklany pręt i szpatułka;
- Żarówka 250 W.
Odczynniki.
- Kwas 3-indolopropionowy (1), 94,6 g (0,5 mol);
- 9,5 l wody destylowanej (H2O);
- ~400 g wodorotlenku sodu (NaOH);
- 116 g niklu Raneya (Ni);
- 1050 mL stężonego kwasu chlorowodorowego (HCl);
- 2 ml kwasu siarkowego (H2SO4 stęż.);
- 210 mL 12N roztwór wodorotlenku sodu (NaOH) aq;
- 180 mL chlorku benzoilu;
- ~1,5 l metanolu (MeOH);
- ~1,6 l etanolu (EtOH);
- 201,2 ml chlorku tionylu (SOCl2);
- 1950 mL disiarczek węgla (CS2);
- 240 g chlorku glinu (AlCl3);
- 2,5 l benzenu;
- 500 ml 2N wodorotlenku sodu (NaOH);
- ~3,2 l eteru dietylowego (Et2O);
- 3,3 l lodowatego kwasu octowego (AcOH);
- 352 g (1,1 mol) bromowodorku pirydyny;
- 5 L Chloroform (CHCl3);
- ~1000 g siarczanu magnezu (MgSO4);
- 307 g (2,35 mol) Ketal etylenowy metyloaminoacetonu (5);
- 4,5 l Benzen;
- ~500 g węgla aktywnego (C);
- ~1 l acetonu;
- ~500 g wodorowęglanu sodu (NaHCO3);
- 80 ml zimnego bezwodnika octowego (Ac2O);
- 1,5 g borohydryku sodu (NaBH4);
- 75 ml dwutlenku siarki (SO2 w płynie);
- 40 g cyjanku sodu (proszek NaCN);
- 300 mL cyjanowodoru (ciecz HCN);
- 78 mL 1,5% roztwór wodorotlenku potasu (KOH) aq;
- 8,5 g uwodnionego arsenianu sodu;
- ~ 50 mL ksylenu;
- 100 ml rozcieńczonego roztworu wodorotlenku amonu (NH4OH);
- 16,9 g metanolanu sodu (MeONa).
Procedura
1-benzoilo-3-(beta-karboksyetylo)-2,3-dihydroindol (2)Kwas 3-indolopropionowy (1), 94,6 g (0,5 mol), rozpuszczono w 600 ml wody zawierającej 20 g wodorotlenku sodu. Roztwór zmieszano z około 100 g katalizatora niklowego Raneya i uwodorniono w temperaturze RT w stalowej bombie uwodorniającej o pojemności 2-3 l pod ciśnieniem 3000-4000 psi (207-276 barów) H2. Redukcja była zwykle zakończona w ciągu 20-30 godzin, po czym katalizator filtrowano i przemywano niewielką ilością wody. Do przesączu dodano 85 ml stężonego kwasu HCl i roztwór schłodzono. Jeśli redukcja była niekompletna, nieprzereagowany kwas indolopropionowy oddzielił się w tym momencie i został usunięty przez filtrację. Przesącz został następnie poddany benzoilowaniu zgodnie ze zwykłą procedurą Schottena-Baumanna, przy użyciu 210 ml 12N wodorotlenku sodu i 180 ml chlorku benzoilu. Roztwór utrzymywano w stanie zasadowym przez cały czas benzoilowania, a temperaturę utrzymywano poniżej 40°C poprzez chłodzenie. Po całkowitym przereagowaniu chlorku benzoilu mieszaninę schłodzono i zakwaszono 300 ml stężonego kwasu HCl. Surowy produkt przefiltrowano i przemyto wodą, po czym ekstrahowano 4 x 1 l gorącej wody w celu usunięcia kwasu benzoesowego. Gorący syropowaty produkt (2), po dekantacji wodnego ekstraktu, wykrystalizowano z kilku objętości metanolu; wydajność 103 g (70%), MP: 151-153 °C.
1-Benzoilo-5-keto-1,2,2a,3,4,5-heksahydrobenz-[cd]-indol (3)
1-Benzoilo-3-(beta-karboksyetylo)-2,3-dihydroindol (2 ), 118 g (0,4 mol), zmieszano z 200 ml czystego chlorku tionylu. Roztwór pozostawiono na 30 minut, po czym delikatnie ogrzewano przez 15-20 minut na łaźni parowej. Nadmiar chlorku tionylu odparowano całkowicie w próżni poniżej 30°C, a surowy chlorek kwasu rozpuszczono w 200 ml disiarczku węgla. Roztwór chlorku kwasu dodano następnie cienkim strumieniem do energicznie mieszanej zawiesiny 240 g chlorku glinu w 1750 ml disiarczku węgla w kolbie o pojemności 5 l (w HOOD!!!). Kompleks oddzielił się, a mieszanie stało się trudne. Mieszaninę ogrzewano pod chłodnicą zwrot ną i mieszano przez godzinę w celu dokończenia reakcji, po czym bardzo ostrożnie rozkładano przez dodanie 500 g lodu, 250 ml stężonego kwasu HCl i 500 ml wody. Podczas rozkładu utrzymywano mieszanie i chłodzenie przez okresową destylację disiarczku węgla in vacuo, a produkt ekstrahowano 2 l benzenu. Ekstrakt dokładnie przemyto 500 ml 2N wodorotlenku sodu w trzech porcjach, a następnie wodą. Wysuszono nad siarczanem magnezu, a następnie odparowano do małej objętości w próżni. Powolne dodanie kilku objętości eteru spowodowało krystalizację żółtego ketonu (3) . Został on przefiltrowany i przemyty eterem; wydajność 85,3 g (77%), MP: 146-147 °C. Próbkę rekrystalizowano do analizy z eteru benzenowego.
1-Benzoilo-3-(beta-karboksyetylo)-2,3-dihydroindol (2 ), 118 g (0,4 mol), zmieszano z 200 ml czystego chlorku tionylu. Roztwór pozostawiono na 30 minut, po czym delikatnie ogrzewano przez 15-20 minut na łaźni parowej. Nadmiar chlorku tionylu odparowano całkowicie w próżni poniżej 30°C, a surowy chlorek kwasu rozpuszczono w 200 ml disiarczku węgla. Roztwór chlorku kwasu dodano następnie cienkim strumieniem do energicznie mieszanej zawiesiny 240 g chlorku glinu w 1750 ml disiarczku węgla w kolbie o pojemności 5 l (w HOOD!!!). Kompleks oddzielił się, a mieszanie stało się trudne. Mieszaninę ogrzewano pod chłodnicą zwrot ną i mieszano przez godzinę w celu dokończenia reakcji, po czym bardzo ostrożnie rozkładano przez dodanie 500 g lodu, 250 ml stężonego kwasu HCl i 500 ml wody. Podczas rozkładu utrzymywano mieszanie i chłodzenie przez okresową destylację disiarczku węgla in vacuo, a produkt ekstrahowano 2 l benzenu. Ekstrakt dokładnie przemyto 500 ml 2N wodorotlenku sodu w trzech porcjach, a następnie wodą. Wysuszono nad siarczanem magnezu, a następnie odparowano do małej objętości w próżni. Powolne dodanie kilku objętości eteru spowodowało krystalizację żółtego ketonu (3) . Został on przefiltrowany i przemyty eterem; wydajność 85,3 g (77%), MP: 146-147 °C. Próbkę rekrystalizowano do analizy z eteru benzenowego.
1-Benzoyl-4-bromo-5-keto-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (4)
1-benzoilo-2,2a,3,4-tetrahydro-4-[metylo-(2-metylo-1,2-dioksolan-2-ylo-metylo)-amino]-benz-[cd]-indol-5-(1H)-on (6)
Roztwór 270 g (0,76 mol) 1-benzoilo-4-bromo-5-keto-1,2,2a,3,4,5-heksahydrobenz-[cd]-indolu(4) i 307 g (2.35 mol) ketalu etylenowego metyloaminoacetonu (5) w 4500 ml suchego benzenu refluksowano pod azotem przez 21 h w 10 l RBF z chłodnicą zwrotną. Mieszaninę schłodzono i przefiltrowano 151 g (93,5%) bromowodorku metyloaminoacetonoetylenoketalu, MP: 158-159 °C.
Przesącz przemyto kilkakrotnie wodą z lodem, po czym ekstrahowano 2,5 l zimnego, rozcieńczonego kwasu HCl zawierającego 150 ml stężonego kwasu. Ekstrakty kwasowe natychmiast dodano do nadmiaru lodowatego, rozcieńczonego wodorotlenku sodu. Produkt ekstrahowano 1 l chloroformu, a roztwór chloroformu suszono nad siarczanem magnezu, traktowano węglem i zatężano w próżni. Pozostały ketal-keton (6) wykrystalizowano z acetonu; MP: i mieszanina MP: 135-136 °C, wydajność 220 g (71 %).
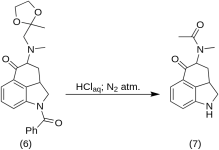
5-Keto-4-[N-metylo-N-acetonyloamino]-1,2,2a,3,4,5-heksahydrobenz-[cd]-indol (7)
20 g 1-benzoilo-2,2a,3,4-tetrahydro-4-[metylo-(2-metylo-1,3-dioksolan-2-ylo-metylo)-amino]-benz-[cd]-indol-5-(1H)-on (6 ) rozpuszczono w mieszaninie 250 ml stężonego kwasu HCl i 250 ml wody, a roztwór utrzymywano pod azotem w temperaturze 37°C w 3-5 l RBF przez pięć dni. Mieszaninę schłodzono, poddano działaniu węgla, przefiltrowano, a przesącz zatężono w próżni do małej objętości. Pozostałość potraktowano nadmiarem wodorowęglanu sodu; produkt ekstrahowano zimnym chloroformem, a rozpuszczalnik usunięto w próżni w temperaturze RT. Surowy diketon (7) sproszkowano, przesączono około 75 ml eteru benzenowego 1:1 i przefiltrowano; wydajność 9,8 g (77%), MP: 105-107 °C. Próbkę do analizy rekrystalizowano z eteru benzenowego lub etanolu; MP: 109-110 °C; monochlorowodorek otrzymano z rozcieńczonego etanolu; MP: 200 °C dec.
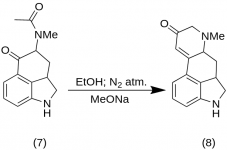
9-Keto-7-metylo-4,5,5a,6,6a,7,8,9-oktahydroindolo[4,3-fg]-chinolina (8)
25 g 5-Keto-4-[N-metylo-N-acetonylo]-amino-1,2,2a,3,4,5-heksahydrobenz-[cd]-indolu (7) zmieszano z 550 ml etanolu absolutnego. Mieszaninę mieszano pod azotem i schłodzono do -15 °C w 2-5 L RBF. Następnie dodano 16,9 g metanolanu sodu i mieszano mieszaninę w temperaturze od -10°C do -12°C przez dziesięć minut. Mieszaninę reakcyjną schłodzono do -25 °C, a produkt przefiltrowano na 6,5-calowym lejku Buchnera i przemyto niewielką ilością zimnego etanolu i eteru. Przy minimalnej ekspozycji na powietrze (zawiera metatlenek sodu!) surowy keton (8) został natychmiast przesączony niewielką ilością lodowatej wody i ponownie przefiltrowany. Przemyto wodą z lodem, etanolem i eterem; wydajność 16,2 g (69%), MP: 145-147 °C. Próbkę analityczną rekrystalizowano z rozcieńczonego etanolu; MP: 155-157 °C; Dichlorowodorek przygotowano i rekrystalizowano z wodnego acetonu; MP: 270 °C dec.
4-Acetylo-9-keto-7-metylo-4,5,5a,6,6a,7,8,9-oktahydroindolo[4,3-fg]-chinolina (9)
9-Keto-7-metylo-4,5,5a,6,6a,7,8,9-oktahydroindolo[4,3-fg]-chinolina (8), 24 g, została dodana do 80 ml zimnego bezwodnika octowego. Mieszaninę utrzymywano w temperaturze 25°C w 200 ml RBF przez około 5 minut, po czym dokładnie schłodzono, a produkt (9) przefiltrowano i przemyto eterem; wydajność 20,5 g (76%), mp: 167-170 °C. Drugi zbiór otrzymano przez odparowanie filtratu; zwiększyło to całkowitą wydajność do 82%. Próbkę rekrystalizowano z acetonu-etanolu; MP: 169-170 °C; Chlorowodorek przygotowano w etanolu i rekrystalizowano z wodnego etanolu; MP: 250 °C dec.
9-Keto-7-metylo-4,5,5a,6,6a,7,8,9-oktahydroindolo[4,3-fg]-chinolina (8), 24 g, została dodana do 80 ml zimnego bezwodnika octowego. Mieszaninę utrzymywano w temperaturze 25°C w 200 ml RBF przez około 5 minut, po czym dokładnie schłodzono, a produkt (9) przefiltrowano i przemyto eterem; wydajność 20,5 g (76%), mp: 167-170 °C. Drugi zbiór otrzymano przez odparowanie filtratu; zwiększyło to całkowitą wydajność do 82%. Próbkę rekrystalizowano z acetonu-etanolu; MP: 169-170 °C; Chlorowodorek przygotowano w etanolu i rekrystalizowano z wodnego etanolu; MP: 250 °C dec.
4-acetylo-9-hydroksy-7-metylo-4,5,5a,6,6a,7,8,9-oktahydroindolo[4,3-fg]-chinolina (10)
10 g 4-acetylo-9-keto-7-metylo-4,5,5a,6,6a,7,8,9-oktahydroksyindolo-[4,3-fg]-chinoliny (9 ) dodano do mieszaniny 150 ml metanolu i 10 ml wody w 500 ml RBF. Dodano 1,5 g borohydryku sodu i pozostawiono reakcję w temperaturze RT do małej objętości, a następnie dodano mieszaninę 15 ml stężonego kwasu HCl i 60 ml wody. Chlorowodorek (10) , który oddzielił się po schłodzeniu, przefiltrowano i przemyto metanolem, uzyskując 9,0 g (79%). Próbkę rekrystalizowano z rozcieńczonego etanolu; MP: 245-246 °C dec.
Chlorowodorek 4-acetylo-9-chloro-7-metylo-4,5,5a,6,6a,7,8,9-oktahydroindolo[4,3-fg]-chinoliny (11)
Chlorowodorek 4-acetylo-9-hydroksy-7-metylo-4,5,5a,6,6a,7,8,9-oktahydroindolo[4,3-fg]-chinoliny (10), 3.1 g, rozpuszczono w 75 ml ciekłego dwutlenku siarki zawartego w szklanej powłoce w stalowym autoklawie o pojemności 500 ml. Dodano 1,2 ml chlorku tionylu, naczynie zamknięto i utrzymywano w temperaturze 25°C przez 6 h. Autoklaw odpowietrzono, a mieszaninę reakcyjną usunięto. Dwutlenek siarki odparowano, a objętość roztworu utrzymywano na stałym poziomie przez powolne dodawanie suchego eteru. Amorficzny chlorowodorek chloru (11) przefiltrowano, przemyto eterem i wysuszono w próżni, MP: 130-135 °C dec. Wydajność 3,5 g. Użycie alkoholu 9-beta-epimerycznego w tej reakcji dało ten sam chlorek z porównywalną wydajnością.
4-Acetylo-9-cyjano-7-metylo-4,5,5a,6,6a,7,8,9-oktahydroindolo[4,3-fg]-chinolina (12)
Suchy, sproszkowany cyjanek sodu, 40 g, dodano do 300 ml lodowatego ciekłego cyjanowodoru. Mieszaninę mieszano i chłodzono w lodzie, po czym dodano 7,5 g surowego amorficznego chlorowodorku 4-acetylo-9-chloro-7-metylo-4,5,5a,6,6a,7,8,9-oktahydroindolo[4,3f/g]-chinoliny (11 ) . Mieszanie kontynuowano w 500 ml RBF przez 30 minut, po czym cyjanowodór szybko oddestylowano pod zmniejszonym ciśnieniem poniżej około 10°C. Pozostałość zmieszano z chloroformem i wodą z lodem, a powstałą mieszaninę przefiltrowano. Warstwę organiczną oddzielono, a fazę wodną ekstrahowano dwukrotnie chloroformem. Połączone ekstrakty wysuszono nad siarczanem magnezu, odbarwiono, a rozpuszczalnik oddestylowano in vacuo. Produkt (12) wykrystalizowano z octanu etylu; wydajność 3,3 g (54% w stosunku do chlorowodorku alkoholu), m.p. 172-174 °C. Rekrystalizacja z tego samego rozpuszczalnika podniosła m.p. do 181-182 °C.
Suchy, sproszkowany cyjanek sodu, 40 g, dodano do 300 ml lodowatego ciekłego cyjanowodoru. Mieszaninę mieszano i chłodzono w lodzie, po czym dodano 7,5 g surowego amorficznego chlorowodorku 4-acetylo-9-chloro-7-metylo-4,5,5a,6,6a,7,8,9-oktahydroindolo[4,3f/g]-chinoliny (11 ) . Mieszanie kontynuowano w 500 ml RBF przez 30 minut, po czym cyjanowodór szybko oddestylowano pod zmniejszonym ciśnieniem poniżej około 10°C. Pozostałość zmieszano z chloroformem i wodą z lodem, a powstałą mieszaninę przefiltrowano. Warstwę organiczną oddzielono, a fazę wodną ekstrahowano dwukrotnie chloroformem. Połączone ekstrakty wysuszono nad siarczanem magnezu, odbarwiono, a rozpuszczalnik oddestylowano in vacuo. Produkt (12) wykrystalizowano z octanu etylu; wydajność 3,3 g (54% w stosunku do chlorowodorku alkoholu), m.p. 172-174 °C. Rekrystalizacja z tego samego rozpuszczalnika podniosła m.p. do 181-182 °C.
9-karbometoksy-7-metylo-4,5,5a,6,6a,7,8,9-oktahydroindolo[4,3-fg]-chinolina (13)
Powyższy produkt (12) , 1,0 g, zmieszano z 15 ml metanolu i 0,25 ml wody. Mieszaninę schłodzono i powoli dodano 2 ml stężonego kwasu siarkowego. Roztwór zamknięto w szklanej probówce pod azotem i ogrzewano w temperaturze 100°C przez 23-24 h w 100 ml RBF z chłodnicą zwrotną. Mieszaninę poddano działaniu odbarwionego węgla, a następnie zatężono w próżni do około 10 ml. Przelano ją do mieszaniny chloroformu (30 mL), lodu i 10 g wodorowęglanu sodu. Warstwę chloroformu oddzielono, a fazę wodną ekstrahowano 3 x 10 ml porcjami chloroformu. Połączone ekstrakty wysuszono nad siarczanem magnezu, odparowano do sucha, a produkt (13) wykrystalizowano z benzenu; wydajność 0,51 g (53%), MP: 159-160 °C. Produktrekrystalizowano z octanu etylu; MP: 160-161 °C.
Powyższy produkt (12) , 1,0 g, zmieszano z 15 ml metanolu i 0,25 ml wody. Mieszaninę schłodzono i powoli dodano 2 ml stężonego kwasu siarkowego. Roztwór zamknięto w szklanej probówce pod azotem i ogrzewano w temperaturze 100°C przez 23-24 h w 100 ml RBF z chłodnicą zwrotną. Mieszaninę poddano działaniu odbarwionego węgla, a następnie zatężono w próżni do około 10 ml. Przelano ją do mieszaniny chloroformu (30 mL), lodu i 10 g wodorowęglanu sodu. Warstwę chloroformu oddzielono, a fazę wodną ekstrahowano 3 x 10 ml porcjami chloroformu. Połączone ekstrakty wysuszono nad siarczanem magnezu, odparowano do sucha, a produkt (13) wykrystalizowano z benzenu; wydajność 0,51 g (53%), MP: 159-160 °C. Produktrekrystalizowano z octanu etylu; MP: 160-161 °C.
Syntetyczny kwas dl-lizergowy (14)
Mieszaninę 9-karbometoksy-7-metylo-4,5,5a,6,6a,7,8,9-oktahydroindolo-[4,3-fg]-chinoliny (13), 3,9 g, i 78 ml 1,5% roztworu wodorotlenku potasu poddano refluksowi przez 30 minut pod azotem. Dodano uwodniony arsenian sodu (8,5 g) i nikiel Raneya (16 g, mokry), uprzednio dezaktywowany przez gotowanie w zawiesinie ksylenu, a mieszaninę ogrzewano pod chłodnicą zwrotną i mieszano w atmosferze azotu przez 20 godzin w 200 ml RBF z chłodnicą zwrotną. Roztwór poddano działaniu węgla, a surowy kwas lizergowy (14 ) wytrącono przez neutralizację do pH 5,6. Został on przefiltrowany i przemyty wodą; wydajność 1,04 g, MP: 240-242 °C dec. Uzyskano również drugi zbiór, 0,16 g, MP: 233-235 °C dec.; całkowita wydajność 30%. Kwas można było oczyścić rozpuszczając go w rozcieńczonym wodorotlenku amonu, traktując węglem odbarwiającym i ponownie wytrącając dwutlenkiem węgla, MP: 242-243 ° C dec; mieszanina z kwasem dl-lizergowym wytworzonym z naturalnego kwasu d-lizergowego również wynosiła 242-243 ° C dec. Bezwodny kwas otrzymano przez suszenie w próżni przez kilka godzin w temperaturze 150 ° C.
Mieszaninę 9-karbometoksy-7-metylo-4,5,5a,6,6a,7,8,9-oktahydroindolo-[4,3-fg]-chinoliny (13), 3,9 g, i 78 ml 1,5% roztworu wodorotlenku potasu poddano refluksowi przez 30 minut pod azotem. Dodano uwodniony arsenian sodu (8,5 g) i nikiel Raneya (16 g, mokry), uprzednio dezaktywowany przez gotowanie w zawiesinie ksylenu, a mieszaninę ogrzewano pod chłodnicą zwrotną i mieszano w atmosferze azotu przez 20 godzin w 200 ml RBF z chłodnicą zwrotną. Roztwór poddano działaniu węgla, a surowy kwas lizergowy (14 ) wytrącono przez neutralizację do pH 5,6. Został on przefiltrowany i przemyty wodą; wydajność 1,04 g, MP: 240-242 °C dec. Uzyskano również drugi zbiór, 0,16 g, MP: 233-235 °C dec.; całkowita wydajność 30%. Kwas można było oczyścić rozpuszczając go w rozcieńczonym wodorotlenku amonu, traktując węglem odbarwiającym i ponownie wytrącając dwutlenkiem węgla, MP: 242-243 ° C dec; mieszanina z kwasem dl-lizergowym wytworzonym z naturalnego kwasu d-lizergowego również wynosiła 242-243 ° C dec. Bezwodny kwas otrzymano przez suszenie w próżni przez kilka godzin w temperaturze 150 ° C.
Attachments
Last edited: