
G.Patton
Expert
- Joined
- Jul 5, 2021
- Messages
- 2,727
- Solutions
- 3
- Reaction score
- 2,886
- Points
- 113
- Deals
- 1

Introduction
Lysergic acid, the basic fragment derived from the ergot alkaloids, has been synthesized in a fourteen sequence beginning with 3-beta-carboxyethylindole. The starting material was converted to the intermediate 1-benzoyl-5-keto-1,2,2a-3,4,5-hexahydrobenz-[cd]-indole (3), containing three of the four rings present in lysergic acid. This ketone in turn was transformed into the tetracyclic compound, 9-keto-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4.3-fe]-quinoline (8), and thence to Lysergic acid (14). This synthesis isn't simple and takes a lot of laboratory experience and chemistry knowledge. Moreover, there are several manipulations with hazardous substances, which have to be carried out with strict safety measures.
Boiling Point: 536.2±50.0 °C at 760 mm Hg;
Melting Point: 240 °C;
Molecular Weight: 268.31 g/mole;
Density: 1.4 ± 0.1 g/mL;
CAS Number: 82-58-6.
Equipment and glassware:

- Steel hydrogenation reactor 2-3 L;
- Steel autoclave 500 mL;
- Laboratory scale (0.01 — 500 g is suitable);
- Round bottom flasks 100, 200, 500 mL, 5 and 10 L;
- Hydrogen (H2) compressor and origin;
- Buchner flask and funnel (large) 5 L [Schott filter may be used for small quantities];
- Rotovap machine (large);
- Vacuum source;
- Separatory funnels 500 mL and 2 L;
- Nitrogen balloon ~50-70 L (1 bar);
- Septum caps for flasks;
- Salted ice water bath;
- 5 L x2, 2 L x2; 1 L x2; 500 mL x2; 100 mL x3; 50 mL x2 Beakers;
- Glass syringe or Pasteur pipette;
- Magnetic stirrer or top stirrer;
- Vacuum distillation setup;
- Reflux condenser;
- Retort stand and clamp for securing apparatus;
- Laboratory grade thermometer (-20 °C to 200 °C) with flask adapter;
- pH Indicator paper;
- Glass rod and spatula;
- 250 watt bulb.
Reagents:
- 3-Indolepropionic acid (1), 94.6 g (0.5 mol);
- 9.5 L Distilled water (H2O);
- ~400 g Sodium hydroxide (NaOH);
- 116 g Raney nickel (Ni);
- 1050 mL Hydrochloric (HCl) acid concentrated;
- 2 mL Sulfuric acid (H2SO4 conc.);
- 210 mL 12N Sodium hydroxide (NaOH) aq solution;
- 180 mL Benzoyl chloride;
- ~1.5 L Methanol (MeOH);
- ~1.6 L Ethanol (EtOH);
- 201.2 mL Thionyl chloride (SOCl2);
- 1950 mL Carbon disulfide (CS2);
- 240 g Aluminum chloride (AlCl3);
- 2.5 L Benzene;
- 500 mL 2N Sodium hydroxide (NaOH);
- ~3.2 L Diethyl ether (Et2O);
- 3.3 L Glacial acetic acid (AcOH);
- 352 g (1.1 mol) Pyridine hydrobromide perbromide;
- 5 L Chloroform (CHCl3);
- ~1000 g Magnesium sulfate (MgSO4);
- 307 g (2.35 mol) Methylaminoacetone ethylene ketal (5);
- 4.5 L Benzene;
- ~500 g Activated carbon (C);
- ~1 L Acetone;
- ~500 g Sodium bicarbonate (NaHCO3);
- 80 mL Cold acetic anhydride (Ac2O);
- 1.5 g Sodium borohydride (NaBH4);
- 75 mL Sulfur dioxide (SO2 liquid);
- 40 g Sodium cyanide (NaCN powder);
- 300 mL Hydrogen cyanide (HCN liquid);
- 78 mL 1.5% Potassium hydroxide (KOH) aq. solution;
- 8.5 g Hydrated sodium arsenate;
- ~ 50 mL Xylene;
- 100 mL Ammonium hydroxide (NH4OH) dilute solution;
- 16.9 g Sodium methoxide (MeONa).
Procedure
1-Benzoyl-3-(beta-carboxyethyl)-2,3-dihydroindole (2)3-Indolepropionic acid (1), 94.6 g (0.5 mol), was dissolved in 600 mL of water containing 20 g of sodium hydroxide. The solution was mixed with about 100 g of Raney nickel catalyst and hydrogenated at RT in a 2-3 L steel hydrogenation bomb at 3000-4000 psi (207-276 bar) H2 pressure. Reduction was usually complete in 20-30 h, after which the catalyst was filtered and washed with a little water. Concentrated HCl acid, 85 mL, was added to the filtrate, and the solution was cooled. If the reduction was incomplete, unreacted indolepropionic acid separated at this point and was removed by filtration. The filtrate was then benzoylated by the usual Schotten-Baumann procedure, using 210 mL of 12N sodium hydroxide and 180 mL of benzoyl chloride. The solution was kept alkaline throughout the benzoylation, and the temperature was kept below 40 °C by cooling. When the benzoyl chloride was completely reacted, the mixture was cooled and acidified with 300 mL of concentrated HCl acid. The crude product was filtered and washed with water, after which it was extracted with 4 x 1 L portions of hot water to remove the benzoic acid. The hot syrupy product (2), after decantation of the aqueous extract, was crystallized from a few volumes of methanol; yield 103 g (70 %), MP: 151-153 °C.
1-Benzoyl-5-keto-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (3)
1-Benzoyl-3-(beta-carboxyethyl)-2,3-dihydroindole (2), 118 g (0.4 mol), was mixed with 200 mL of pure thionyl chloride. The solution was allowed to stand for 30 min, after which it was warmed gently for 15-20 min on the steam-bath. Excess thionyl chloride was evaporated completely below 30 °C in vacuo, and the crude acid chloride was dissolved in 200 mL of carbon disulfide. The solution of the acid chloride was then added in a thin stream to a vigorously stirred suspension of 240 g of aluminum chloride in 1750 mL of carbon disulfide contained in a 5 L flask (in HOOD!!!). A complex separated, and stirring became difficult. The mixture was heated under reflux and stirred for one hour to complete the reaction, after which it was decomposed very carefully by adding 500 g of ice, 250 mL of conc. HCl acid and 500 mL of water. During the decomposition, stirring was maintained, and cooling was affected by periodic distillation of the carbon disulfide in vacuo, and the product was extracted with 2 L of benzene. The extract was washed thoroughly with 500 mL of 2N sodium hydroxide in three portions and then with water. It was dried over magnesium sulfate and then evaporated to small volume in vacuo. Slow addition of several volumes of ether caused the yellow ketone (3) to crystallize. It was filtered and washed with ether; yield 85.3 g (77 %), MP: 146-147 °C. A sample was recrystallized for analysis from benzene-ether.
1-Benzoyl-3-(beta-carboxyethyl)-2,3-dihydroindole (2), 118 g (0.4 mol), was mixed with 200 mL of pure thionyl chloride. The solution was allowed to stand for 30 min, after which it was warmed gently for 15-20 min on the steam-bath. Excess thionyl chloride was evaporated completely below 30 °C in vacuo, and the crude acid chloride was dissolved in 200 mL of carbon disulfide. The solution of the acid chloride was then added in a thin stream to a vigorously stirred suspension of 240 g of aluminum chloride in 1750 mL of carbon disulfide contained in a 5 L flask (in HOOD!!!). A complex separated, and stirring became difficult. The mixture was heated under reflux and stirred for one hour to complete the reaction, after which it was decomposed very carefully by adding 500 g of ice, 250 mL of conc. HCl acid and 500 mL of water. During the decomposition, stirring was maintained, and cooling was affected by periodic distillation of the carbon disulfide in vacuo, and the product was extracted with 2 L of benzene. The extract was washed thoroughly with 500 mL of 2N sodium hydroxide in three portions and then with water. It was dried over magnesium sulfate and then evaporated to small volume in vacuo. Slow addition of several volumes of ether caused the yellow ketone (3) to crystallize. It was filtered and washed with ether; yield 85.3 g (77 %), MP: 146-147 °C. A sample was recrystallized for analysis from benzene-ether.
1-Benzoyl-4-bromo-5-keto-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (4)
1-Benzoyl-2,2a,3,4-tetrahydro-4-[methyl-(2-methyl-1,2-dioxolan-2-yl-methyl)-amino]-benz-[cd]-indol-5-(1H)-one (6)
A solution of 270 g (0.76 mol) of 1-benzoyl-4-bromo-5-keto-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole(4) and 307 g (2.35 mol) of methylaminoacetone ethylene ketal (5) in 4500 mL of dry benzene was refluxed under nitrogen for 21 h in 10 L RBF with reflux condenser. The mix was cooled, and 151 g (93.5 %) of methylaminoacetone ethylene ketal hydrobromide was filtered, MP: 158-159 °C.
The filtrate was washed several times with ice-water, after which it was extracted with 2.5 L of cold, dilute HCl acid containing 150 mL of the concentrated acid. The acid extracts were immediately added to an excess of ice-cold dilute sodium hydroxide. The product was extracted with 1 L of chloroform, and the chloroform solution was dried over magnesium sulfate, treated with carbon and concentrated in vacuo. The residual ketal-ketone (6) was crystallized from acetone; MP: and mixture MP: 135-136 °C, yield 220 g (71 %).
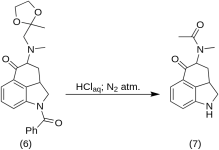
5-Keto-4-[N-methyl-N-acetonylamino]-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (7)
20 g of 1-benzoyl-2,2a,3,4-tetrahydro-4-[methyl-(2-methyl-1,3-dioxolan-2-yl-methyl)-amino]-benz-[cd]-indol-5-(1H)-one (6) was dissolved in a mixture of 250 mL of concentrated HCl acid and 250 mL of water, and the solution was kept under nitrogen at 37 °C in 3-5 L RBF for five days. The mixture was cooled, treated with carbon, filtered and the filtrate was concentrated in vacuo to small volume. The residue was treated with excess sodium bicarbonate; the product was extracted with cold chloroform, and the solvent was removed in vacuo at RT. The crude diketone (7) was powdered, slurried with about 75 mL of 1:1 benzene-ether, and filtered; yield 9.8 g (77 %), MP: 105-107 °C. A sample for analysis was recrystallized from benzene-ether or ethanol; MP: 109-110 °C; A monohydrochloride was obtained from dilute ethanol; MP: 200 °C dec.
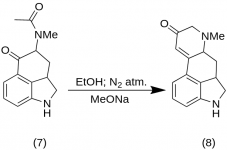
9-Keto-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoline (8)
25 g of 5-Keto-4-[N-methyl-N-acetonyl]-amino-1,2,2a,3,4,5-hexahydrobenz-[cd]-indole (7) was mixed with 550 mL of absolute ethanol. The mix was stirred under nitrogen and cooled to -15 °C in 2-5 L RBF. Sodium methoxide, 16.9 g, was then added, and the mixture was stirred at -10 °C to -12 °C for ten minutes. The reaction mixture was cooled to —25 °C, and the product was filtered on a 6.5-inch buchner funnel and washed with a little cold ethanol and ether. With the very minimum exposure to air (contains sodium methoxide!) the crude ketone (8) was immediately slurried with a little ice-water and refiltered. It was washed with ice-water, ethanol and ether; yield 16.2 g (69 %), MP: 145-147 °C. An analytical sample was recrystallized from dilute ethanol; MP: 155-157 °C; The dihydrochloride was prepared and recrystallized from aqueous acetone; MP: 270 °C dec.
4-Acetyl-9-keto-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoline (9)
9-Keto-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoline (8), 24 g, was added to 80 mL of cold acetic anhydride. The mixture was kept at 25 °C in 200 mL RBF for about 5 min, after which it was thoroughly cooled, and the product (9) was filtered and washed with ether; yield 20.5 g (76 %), mp: 167-170 °C. A second crop was obtained by evaporation of the filtrate; this raised the total yield to 82%. A sample was recrystallized from acetone-ethanol; MP: 169-170 °C; The hydrochloride was prepared in ethanol and was recrystallized from aqueous ethanol; MP: 250 °C dec.
9-Keto-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoline (8), 24 g, was added to 80 mL of cold acetic anhydride. The mixture was kept at 25 °C in 200 mL RBF for about 5 min, after which it was thoroughly cooled, and the product (9) was filtered and washed with ether; yield 20.5 g (76 %), mp: 167-170 °C. A second crop was obtained by evaporation of the filtrate; this raised the total yield to 82%. A sample was recrystallized from acetone-ethanol; MP: 169-170 °C; The hydrochloride was prepared in ethanol and was recrystallized from aqueous ethanol; MP: 250 °C dec.
4-Acetyl-9-hydroxy-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoline (10)
10 g of 4-acetyl-9-keto-7-methyl-4,5,5a,6,6a,7,8,9-octahydroxindolo-[4,3-fg]-quinoline (9) was added to a mixture of 150 mL of methanol and 10 mL of water in 500 mL RBF. Sodium borohydride, 1.5 g was added, and the reaction was allowed to proceed at RT to small volume, and a mix of 15 mL of conc. HCl acid and 60 mL of water was added. The hydrochloride (10) which separated on cooling was filtered and washed with methanol, 9.0 g (79 %). A sample was recrystallized from dilute ethanol; MP: 245-246 °C dec.
4-Acetyl-9-chloro-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoline hydrochloride (11)
4-Acetyl-9-hydroxy-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoline hydrochloride (10), 3.1 g, was dissolved in 75 mL of liquid sulfur dioxide contained in a glass liner in a 500 mL steel autoclave. Thionyl chloride, 1.2 mL was added, and the vessel was sealed and kept at 25 °C for 6 h. The autoclave was vented, and the reaction mix was removed. Sulfur dioxide was allowed to evaporate while the volume of the solution was kept constant by the slow addition of dry ether. The amorphous chloro hydrochloride (11) was filtered, washed with ether, and dried in vacuo, MP: 130-135 °C dec. Yield 3.5 g. Use of the 9-beta-epimeric alcohol in this reaction gave the same chloride in comparable yield.
4-Acetyl-9-cyano-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoline (12)
Dry, powdered sodium cyanide, 40 g., was added to 300 mL. of ice-cold liquid hydrogen cyanide. The mixture was stirred and cooled in ice, and 7.5 g of the crude amorphous 4-acetyl-9-chloro-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo [4,3f/g]-quinoline hydrochloride (11) above was added. Stirring was continued in 500 mL RBF for 30 min, after which the hydrogen cyanide was quickly distilled under reduced pressure below about 10 °C. The residue was mixed with chloroform and ice-water, and the resulting mixture was filtered. The organic layer was separated, and the aqueous phase was extracted twice with chloroform. The combined extracts were dried over magnesium sulfate, decolorized and the solvent was distilled in vacuo. The product (12) was crystallized from ethyl acetate; yield 3.3 g. (54% over-all based on the alcohol hydrochloride), m.p. 172-174 °C. Recrystallization from the same solvent raised the m.p. to 181-182 °C.
Dry, powdered sodium cyanide, 40 g., was added to 300 mL. of ice-cold liquid hydrogen cyanide. The mixture was stirred and cooled in ice, and 7.5 g of the crude amorphous 4-acetyl-9-chloro-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo [4,3f/g]-quinoline hydrochloride (11) above was added. Stirring was continued in 500 mL RBF for 30 min, after which the hydrogen cyanide was quickly distilled under reduced pressure below about 10 °C. The residue was mixed with chloroform and ice-water, and the resulting mixture was filtered. The organic layer was separated, and the aqueous phase was extracted twice with chloroform. The combined extracts were dried over magnesium sulfate, decolorized and the solvent was distilled in vacuo. The product (12) was crystallized from ethyl acetate; yield 3.3 g. (54% over-all based on the alcohol hydrochloride), m.p. 172-174 °C. Recrystallization from the same solvent raised the m.p. to 181-182 °C.
9-Carbomethoxy-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoline (13)
The product (12) just above, 1.0 g, was mixed with 15 mL of methanol and 0.25 mL of water. The mix was cooled and 2 mL of concentrated sulfuric acid was added slowly. The solution was sealed in a glass tube under nitrogen and heated at 100 °C for 23 to 24 h in 100 mL RBF with reflux condenser. The mixture was treated with decolorized carbon and then concentrated in vacuo to about 10 mL. It was poured onto a mix of chloroform (30 mL), ice and 10 g of sodium bicarbonate. The chloroform layer was separated, and the aqueous phase was extracted with 3 x 10 mL portions of chloroform. The combined extracts were dried over magnesium sulfate, evaporated to dryness, and the product (13) was crystallized from benzene; yield 0.51 g (53 %), MP: 159-160 °C. It was recrystallized from ethyl acetate; MP: 160-161 °C;
The product (12) just above, 1.0 g, was mixed with 15 mL of methanol and 0.25 mL of water. The mix was cooled and 2 mL of concentrated sulfuric acid was added slowly. The solution was sealed in a glass tube under nitrogen and heated at 100 °C for 23 to 24 h in 100 mL RBF with reflux condenser. The mixture was treated with decolorized carbon and then concentrated in vacuo to about 10 mL. It was poured onto a mix of chloroform (30 mL), ice and 10 g of sodium bicarbonate. The chloroform layer was separated, and the aqueous phase was extracted with 3 x 10 mL portions of chloroform. The combined extracts were dried over magnesium sulfate, evaporated to dryness, and the product (13) was crystallized from benzene; yield 0.51 g (53 %), MP: 159-160 °C. It was recrystallized from ethyl acetate; MP: 160-161 °C;
Synthetic dl-Lysergic Acid (14)
A mix of 9-carbomethoxy-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoline (13), 3.9 g, and 78 mL of 1.5% potassium hydroxide solution was refluxed for 30 min under nitrogen. Hydrated sodium arsenate, 8.5 g, and Raney nickel (16 g, wet), previously deactivated by boiling in xylene suspension, was added, and the mix was heated under reflux and stirred in a nitrogen atmosphere for 20 hours in 200 mL RBF with reflux condenser. The solution was treated with carbon, and the crude lysergic acid (14) was precipitated by neutralization to pH 5.6. It was filtered and washed with water; yield 1.04 g, MP: 240-242 °C dec. A second crop, 0.16 g, MP: 233-235 °C dec. was also obtained; total yield 30%. The acid could be purified by dissolving it in dilute ammonium hydroxide, treating with decolorizing carbon, and reprecipitating with carbon dioxide., MP: 242-243 °C dec; a mix with dl-lysergic acid made from natural d-lysergic acid was likewise 242-243 °C dec. The anhydrous acid was obtained by drying in vacuo for several hours at 150 °C.
A mix of 9-carbomethoxy-7-methyl-4,5,5a,6,6a,7,8,9-octahydroindolo-[4,3-fg]-quinoline (13), 3.9 g, and 78 mL of 1.5% potassium hydroxide solution was refluxed for 30 min under nitrogen. Hydrated sodium arsenate, 8.5 g, and Raney nickel (16 g, wet), previously deactivated by boiling in xylene suspension, was added, and the mix was heated under reflux and stirred in a nitrogen atmosphere for 20 hours in 200 mL RBF with reflux condenser. The solution was treated with carbon, and the crude lysergic acid (14) was precipitated by neutralization to pH 5.6. It was filtered and washed with water; yield 1.04 g, MP: 240-242 °C dec. A second crop, 0.16 g, MP: 233-235 °C dec. was also obtained; total yield 30%. The acid could be purified by dissolving it in dilute ammonium hydroxide, treating with decolorizing carbon, and reprecipitating with carbon dioxide., MP: 242-243 °C dec; a mix with dl-lysergic acid made from natural d-lysergic acid was likewise 242-243 °C dec. The anhydrous acid was obtained by drying in vacuo for several hours at 150 °C.
Attachments
Last edited: